

Выраженная дилатация правого предсердиякак нетипичный дебют кардиомиопатии,вызванной нарушением в гене LMNA
Авторы: Е.В. Борисова, С.А. Глебова, П.С. Козлов, Е.П. Бурлаченко
АО «Кардиоклиника»; Кузнецовская, 25, Санкт-Петербург, 196105
Е.В. Борисова - доктор медицинских наук, главный врач
С.А. Глебова - кандидат медицинских наук, кардиолог
П.С. Козлов - кандидат медицинских наук, кардиолог
Е.П. Бурлаченко - кандидат медицинских наук, рентгенолог
Участие авторов:
Осмотр пациентки: С.А. Глебова, П.С. Козлов, Е.В. Борисова
Выполнение трансторакальной ЭХО-КГ С.А. Глебова, П.С. Козлов
Выполнение КТ сердца Е.П. Бурлаченко
Написание текста – С.А. Глебова, Е.В. Борисова, П.С. Козлов, Е.П. Бурлаченко
Редактирование – Е.В. Борисова
Мутация гена LMNA, который кодирует два главных ламиновых белка А и С, вызывает целый спектр болезней, называющихся ламинопатией и включающих в себя дилатационную кардиомиопатию, различные типы мышечных дистрофий, липодистрофию, акрогерию и прогерию. Поражение сердечной мышцы при мутациях в гене LMNA в зарубежной литературе описывается как ‘lamin A/C heart disease’. Типичное течение данного заболевания включает в себя следующие этапы: наджелудочковые экстрасистолы, наджелудочковые аритмии, патологию проводящей системы сердца, гипокинетическую кардиомиопатию без дилатации и дилатационную кардиомиопатию. Однако в ряде случаев симптомы могут появляться беспорядочно или в комбинации. Мы представляем случай поздней диагностики ламинопатии, проявившей себя кардиомиопатией, миопатией и парциальной семейной липодистрофией. Особенность кардиомиопатии в том, что при дилатации всех четырёх камер наиболее выражена дилатация правого предсердия. Генетический анализ выявил нарушения в нуклеотидной последовательности в генах LMNA, ANO5, MYPN.
Ключевые слова: ламинопатия, дилатационная кардиомиопатия, аритмии, семейная частичная липодистрофия.
Финансирование. Исследование не имело спонсорской поддержки.
Конфликт интересов. Авторы заявляют об отсутствии конфликта интересов.
Dilatation of the right atrium, as atypical debut of cardiomyopathy caused by lamin A/C (LMNA) mutation.
E.V. Borisova, S.A. Glebova, P.S. Kozlov, E.P.Burlachenko.
Mutation in the LMNA gene, which encodes the two main lamin A and C, cause a spectrum of diseases, called laminopathies, including dilated cardiomyopathy, myopathy, lipodystrophy, akrogeria and progeria. A typical progression lamin A/C heart disease’ might be from premature atrial contractions to atrial arrhythmias, to сardiac сonduction disease , to hypokinetic non- dilated cardiomyopathy and to dilated cardiomyopathy, but features may present ‘out-of-order’ and in combination. We present the case of a late diagnosis of laminopathy, manifestated dilated cardiomyopathy, myopathy and partial lipodystrophy. Cardiomyopathy was presented severe dilatation right atrium with midly dilated another chambers. Genetic testing identified LMNA, ANO5 and MYPN mutation.
История изучения ламинопатий насчитывает уже 20 лет, однако широкий спектр её клинических проявлений часто приводит к несвоевременной диагностике. Ассоциация ламинопатий с высоким риском развития внезапной смерти достигает 46%, что требует пристального внимания клиницистов к данной проблеме [1].
Ядерная ламина это белковая «сетка», выполняющая функцию каркаса ядра клетки, определяющая его размер и форму. Она состоит из четырёх ламиновых белков: А, B1, B2, C, которые способны полимеризироваться и образовывать организованную сеть, лежащую в основе внутренней мембраны ядра в большинстве соматических клеток. Белки А и С кодируются геном LMNA, мутация которого вызывает целый спектр болезней, называющихся ламинопатией, и включающих в себя дилатационную кардиомиопатию, различные типы мышечных дистрофий, липодистрофий, акрогерию и прогерию [2].
В 1999 году французские учёные впервые выявили первые четыре разновидности мутаций в гене LMNA у пациентов, страдающих нейро-мышечной дистрофией Эмери-Дрейфуса. У всех этих пациентов наблюдалась характерная триада симптомов: ранние контрактуры сухожилий, мышечная слабость в плечевых и бедренных группах мышц и дилатационная кардиомиопатия (ДКМП) с нарушениями в проводящей системе сердца. Авторы сделали вывод, что мутации в ядерной ламине являются причиной наследственных мышечных заболеваний [4]. В этом же году американские учёные опубликовали работу, демонстрирующую 5 новых мутаций LMNA гена у 11 пациентов, страдающих мышечной дистрофией Эмери-Дрейфуса аутосомно-доминантного типа. Данные мутации авторы связали с дилатационной кардиомиопатией и поражением проводящей системы сердца у этих пациентов [5]. На сегодняшний день известно несколько сотен мутаций в данном гене.
Поражение сердечной мышцы при мутациях в гене LMNA в зарубежной литературе описывается как ‘lamin A/C heart disease’. Клинические проявления этих мутаций разнообразны от отсутствия видимых кардиологических заболеваний до изолированной дилатации желудочков, аритмогенной кардиомиопатии, гипокинетической кардиомиопатии без дилатации [3]. Безусловно, стадия снижения фракции выброса обычно ассоциирована с дилатацией ЛЖ, но в некоторых ситуациях (например, у носителей мутаций в генах ламина A/C) дилатация ЛЖ может отсутствовать. Именно поэтому в 2016 году эксперты предложили новый промежуточный фенотип ДКМП — гипокинетическую кардиомиопатию без дилатации желудочков - HNDC или DCM [ND-H]. [6]
Заболевания сердца, связанные с патологией ламина А/С относятся к злокачественным, так как проявляются сердечной недостаточностью, часто требующей трансплантации сердца, желудочковыми аритмиями, нарушения проводящей системы, а также сопряжены с высоким риском внезапной смерти [7]. Несмотря на то что болезни сердца, связанные с патологией ламина А/С протекают тяжело и у мужчин, и у женщин, более злокачественные фенотипы и более тяжёлые исходы описаны у мужчин [lamin heart].
Считается, что 10% всех ДКМП обусловлены мутацией в гене LMNA. Поэтому эксперты выделили «красные флаги», которые указывают на генетическую причину заболевания. Например:
- семейные дефекты проводимости (АВ блокады различной степени, внутрижелудочковые блокады);
- скелетно-мышечная семейная патология (мышечная слабость, повышение креатинфосфокиназы);
- жизнеопасные желудочковые тахиаритмии (ЖТА) или семейные случаи внезапной сердечной смерти (ВСС) в возрасте до 45 лет;
- специфические псевдо-инфарктные изменения ЭКГ/ЭХО-КГ в области задне/нижне-боковых сегментов ЛЖ (аномальный Q-зубец/акинезия миокарда) [3].
Пациенты, у которых имеются данные признаки, должны быть своевременно направлены к врачу генетику. В настоящее время польза традиционной медикаментозной терапии ХСН у таких пациентов не изучена. Бета-блокаторы могут улучшить состояние и снизить риск развития жизнеугрожающих аритмий, однако они увеличивают риск развития нарушений проводящей системы сердца. LMNA - один из первых генов, включённых в алгоритм стратификации риска внезапной смерти [8]. Профилактическая имплантация ЭКС может быть полезна у пациентов с прогрессирующими нарушениями проводящей системы сердца и дисфункцией левого желудочка.
Липодистрофия, характерная для мутации в гене LMNA представляет собой семейную частичную липодистрофию типа 2. При данной форме липодистрофии у пациентов нормальное распределение жировой ткани в детстве, а в периоде пубертата жировая ткань исчезает с туловища и конечностей и накапливается области лица и шеи [9].
Пациентка, 28 лет. Жалоб не предъявляла. При плановом профилактическом осмотре было выявлено увеличение тени сердца, в связи с чем рекомендовано выполнение ЭХО-КГ и консультация кардиолога.
При объективном осмотре обращал внимание низкий процент жировой ткани в области туловища и конечностей, выраженная рельефность мускулатуры при незначительном снижении мышечной силы, «широкая шея» (за счёт выраженной аккумуляции жировой ткани в данной области). Складывалось впечатление о псевдогипертрофии мышц плечевого пояса и голеней (рис 1,2). АД 110/70 мм рт ст. ЧСС 48 ударов в минуту. Тоны сердца звучные, слабый систолический шум у основания мечевидного отростка. Дыхание везикулярное, хрипы не выслушиваются. При детальном расспросе пациентка сообщила, что рельефность мускулатуры и редкий пульс отмечается у неё с подросткового возраста. По данному поводу обращались к педиатру, патологии выявлено не было.

Псевдогипертрофия мышц голени.

Типичное перераспределение жировой ткани, аккумуляция в области шеи.
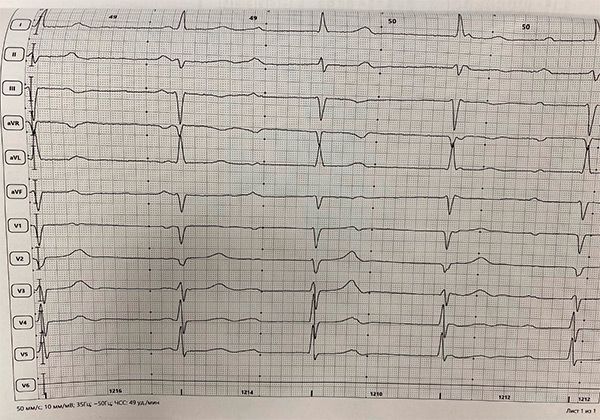
АВ-блокада 3 ст с ЧЖС 49 в минуту.
При выполнении ЭХО-КГ было выявлено значительное увеличение правого предсердия (86х75 мм, 190 мл), умеренное увеличение левого предсердия (передне-задний размер 45 мм), умеренное увеличение правого желудочка, трикуспидальная регургитация 2 степени (рисунок 4). Расчётное давление в ЛА 45 мм рт. ст. Размеры ЛЖ находились на верхней границе нормы. Глобальная сократительная способность ЛЖ сохранена. Митральная регургитация физиологическая. Патологические потоки через межжелудочковую и межпредсердную перегородки не визуализировались. Для уточнения диагноза была проведена компьютерная томография (КТ) сердца.
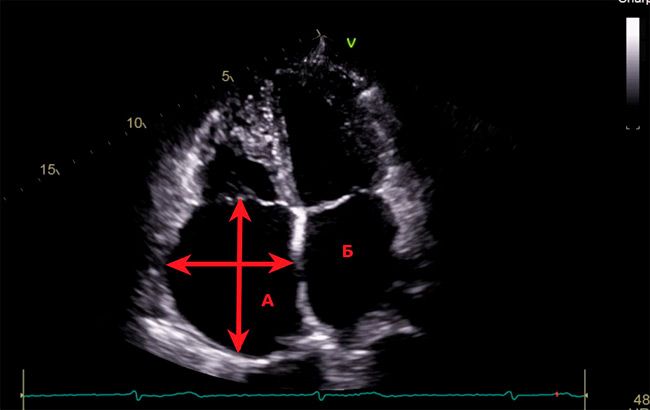
Эхокардиография, 4-х камерная проекция. Выраженная дилатация правого предсердия (86х75 мм). А-правое предсердие. Б-левое предсердие.
На КТ сердца было подтверждено увеличение правого предсердия, его относительные размеры 85х78х93 мм. Остальные камеры увеличены, однако умерено относительно правого предсердия (рис. 5).
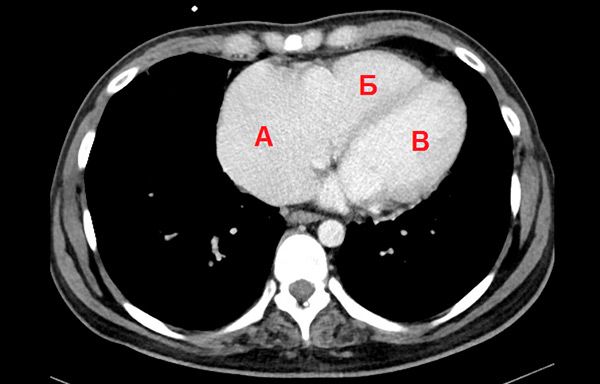
КТ грудной клетки, аксиальная проекция. А-правое предсердие, Б –правый желудочек, В-левый желудочек.
Данных за аномалии лёгочных вен не получено (рис. 6). Для исключения сообщения между полостями сердца был рассчитан показатель Qp-Qs, методом гибридной визуализации. Данный параметр оказался равен 1, что позволило исключить наличие внутрисердечного шунта. Размеры выходных трактов аорты и лёгочной артерии были определены с помощью КТ сердца. Интегралы линейных скоростей через клапаны были получены доплеровским методом при ЭХО-КГ.
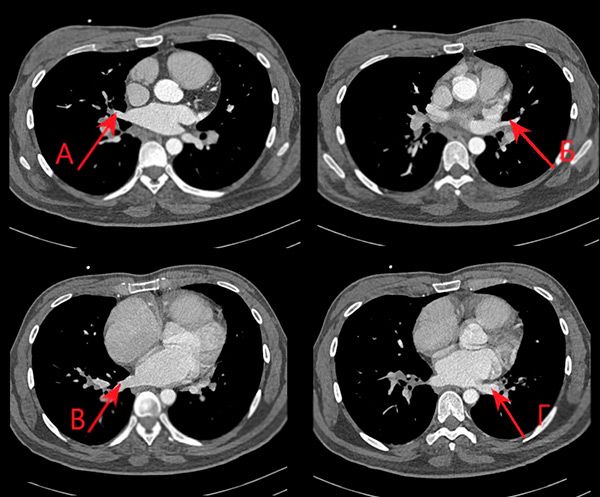
КТ грудной клетки, аксиальная проекция. А-правая нижняя лёгочная вена, Б-левая нижняя лёгочная вена, В правая верхняя лёгочная вена, Г-левая верхняя лёгочная вена.
В биохимическом анализе крови было выявлено повышение КФК до 1521 ЕД/л. При проведении МРТ выявлено увеличение обоих желудочков с сохранением их сократительной способности (КДО ЛЖ 213 мл, КСО ЛЖ 95 мл, ФВ ЛЖ 56%, КДО ПЖ 254 мл, КСО ПЖ 128 мл, ФВ ПЖ 50%). Заключение: МР признаки дилатации всех камер сердца, наиболее выражено правого предсердия. Дополнительно пациентке была выполнена липидограмма, в которой не было выявлено нарушений. В связи с сочетанием идиопатического увеличения правых камер сердца с АВ блокадой 3 степени, с патологией мышц, с нарушением распределения жировой ткани пациентка была направлена на консультацию к генетику. При генетическом анализе были выявлены нарушения в нуклеотидной последовательности в генах LMNA, ANO5, MYPN. Кардиомиопатия, обусловленная мутациями ядерного гена ламина (LMNA), часто ассоциирована с нарушениями сердечного ритма, проводимости и различными скелетно-мышечными расстройствами. Мутации в гене ANO5 служат причиной развития поясно-конечностных мышечных дистрофий. Мутации в гене MYPN являются причиной миопатий. Пациентка была направлена на консультацию к аритмологу для решения вопроса о постановке ЭКС.
Особенностью данного случая является выраженное расширение правого предсердия, при умеренном увеличении остальных камер сердца.
Список литературы:
- van Berlo J.H., de Voogt W.G., van der Kooi A.J., van Tintelen J.P., Bonne G., Yaou R.B. et al. Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death?J. Mol. Med. (Berl). 2005;83(1):79-83.DOI: 10.1007/s00109-004-0589-1
- Charron Р., Arbustini E., Bonne G. What Should the Cardiologist know about Lamin Disease? Arrhythm Electrophysiol Rev. 2012 Sep; 1(1): 22–28. DOI: 10.15420/aer.2012.1.1.22
- Captur G., Arbustini E., Bonne G., Syrris P., Mills K., Wahbi K.. et al. Lamin and the heart. Heart 2018; 104:468–479. DOI:10.1136/heartjnl-2017-312338
- Bonne G., Di Barletta M.R., Varnous S., Bécane H.M., Hammouda E.H., Merlini L. et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nature Genet. 1999; 21: 285–8. DOI: 10.1038/6799
- Fatkin D, MacRae C, Sasaki T. Wolff MR, Porcu M, Frenneaux M, et al. Missense mutations in the rod domain of tle lamin A/C gene as causes of dilated cardiomyopathy and conduction-system disease. N Engl J Med. 1999;341:1715–24. DOI: 10.1056/NEJM199912023412302
- Pinto YM, Elliott PM, Arbustini E, Adler Y, Anastasakis A, Böhm M et al Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non-dilated cardiomyopathy, and its implications for clinical practice: a position statement of the ESC working group on myocardial and pericardial diseases. Eur Heart J. 2016. 14;37(23):1850-8. DOI: 10.1093/eurheartj/ehv727.
- Kayvanpour E., Sedaghat-Hamedani F., Amr A., Lai A., Haas J., Holzer D.B. Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clin Res Cardiol. 2017;106(2):127-139. DOI: 10.1007/s00392-016-1033-6.
- Priori S.G., Blomstro¨m-Lundqvist C., Mazzanti A., Bloma N., Borggrefe M., Camm J. at al, 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac deathDOI:10.1093/eurheartj/ehv316.
- Jeru S., Vatier C., Vantyghem M., Lascols O., Vigouroux C. LMNA-associated partial lipodystrophy: anticipation of metabolic complications. J Med Genet 2017; 0:1–4. DOI:10.1136/jmedgenet-2016-104437
